a multi-task deep learning framework for accurate prediction of microorganism reactive oxygen species scavenging enzymes
Living organisms are constantly exposed to environmental stressors that can cause oxidative damage to their cells and tissues. To alleviate the ROS and induced antagonist effects, microbes rely on the well-evolved ROS-scavenging systems, which is comprised of enzymatic to balance the ROS levels at a steady-state. ROS molecules, including superoxide anion, hydrogen peroxide, hydroxyl radical, and singlet oxygen, are produced due to normal cellular metabolism or exposure to environmental stressors. While these ROS molecules play crucial roles in a variety of cellular processes, their excessive accumulation can lead to oxidative stress and cellular damage. Therefore, microbes have developed diverse defense mechanisms to combat ROS-induced damage, with ROSes playing important roles in scavenging ROS and protecting cells from oxidative damage.
With the rapid advancement of high-throughput sequencing technologies, the volume of genomic and proteomic data generated has increased exponentially, leading to an explosion of sequencing data in recent years . While a multitude of bioinformatics tools and databases exist for protein annotation, such as DEEPARG and ARG_SHINE, these resources still have their own set of limitations , the identification of ROSes still poses a significant challenge. This is due to the complex functional diversity of ROSes proteins and the lack of well-defined sequence motifs that are unique to these proteins. As a result, traditional protein annotation methods have become increasingly inefficient in keeping pace with the exponential growth of sequencing data. This has created a pressing need for the development of fast and efficient computational methods for the accurate annotation of ROSes proteins, which can be accomplished through machine learning and deep learning techniques.
As high-throughput sequencing technologies continue to advance, the amount of protein sequence data is growing exponentially. However, the functions of most proteins still remain unclear. Experimental and computational methods are the two major approaches to determining protein functions. While experimental methods rely on biological experiments to verify protein function, they are much slower than the speed of generating protein sequence data. In contrast, computational methods predict protein functions from protein sequence structures and other information, which is a more efficient and economical approach to determining protein function.
Sequence alignment-based methods, such as BLAST, are widely used for protein functional annotation. However, these methods have limitations in accurately predicting protein function based solely on sequence similarity. For instance, proteins with highly similar sequences may have different functions, while proteins with low sequence similarity may perform similar functions. Additionally, these methods may not be effective in identifying remote homologs or proteins with divergent sequences, leading to incorrect functional annotations. Moreover, sequence alignment-based methods do not consider other critical factors, such as protein structure, post-translational modifications, and protein–protein interactions, which can significantly influence protein function.
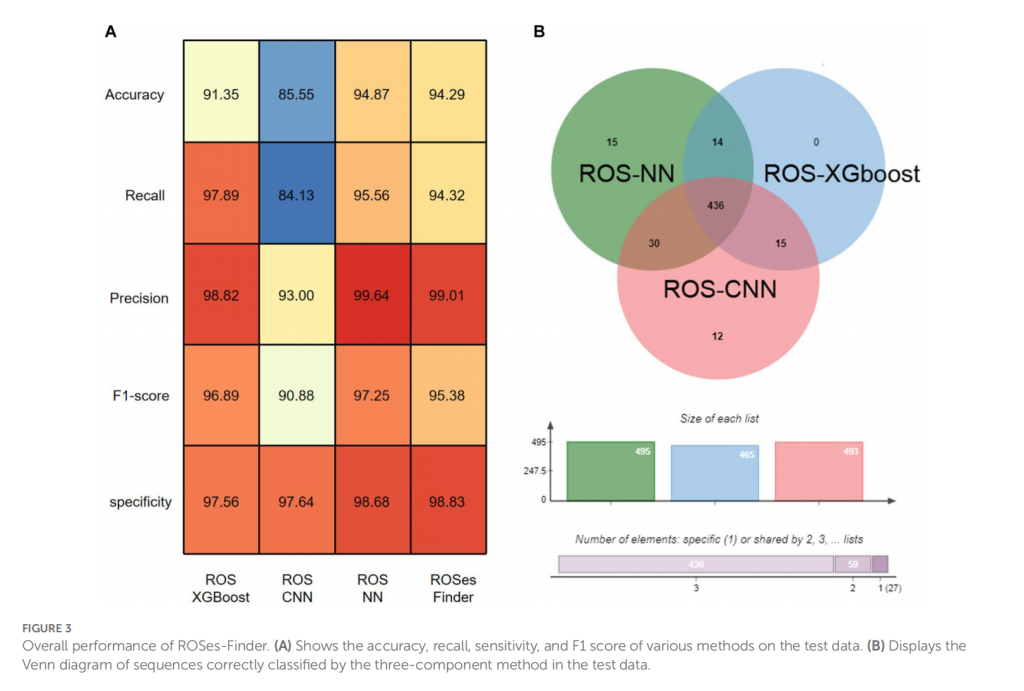
To overcome these limitations, alternative computational methods have been developed, such as machine learning-based approaches. These methods can integrate multiple sources of data, including sequence information, protein structure, and functional annotations from various databases, to improve the accuracy of protein function prediction. By leveraging these methods, we can accelerate the process of determining protein function and unlock the potential of the exponentially growing protein sequence data. This study proposes a new integrated approach called ROSes-FINDER for predicting anti-oxidative protein classes, which overcomes the limitations of existing methods for classifying ROSes. ROSes-FINDER is a multi-task deep learning framework that predicts multiple ROS properties simultaneously, including whether the input protein sequence is ROSes and, if so, what type of ROSes protein it belongs to. To improve the accuracy and robustness of the integrated model, the framework integrates three component methods using a voting-based approach, namely ROSes-CNN, ROSes-NN, and ROSes-XGBOOST. ROSes-CNN uses raw sequence encoding for feature extraction, while ROSes-NN is suitable for predicting protein functions based on sequence information by learning complex non-linear relationships. ROSes-XGBOOST is an ensemble machine learning algorithm that combines the outputs of many decision trees to make a final prediction, making it a useful tool for functional classification. The combination of these three methods can potentially provide more comprehensive and reliable functional predictions for proteins, and the voting-based approach reduces the impact of individual classifier errors, thereby reducing the risk of overfitting.
Website : https://github.com/alienn233/ROSes-Finder